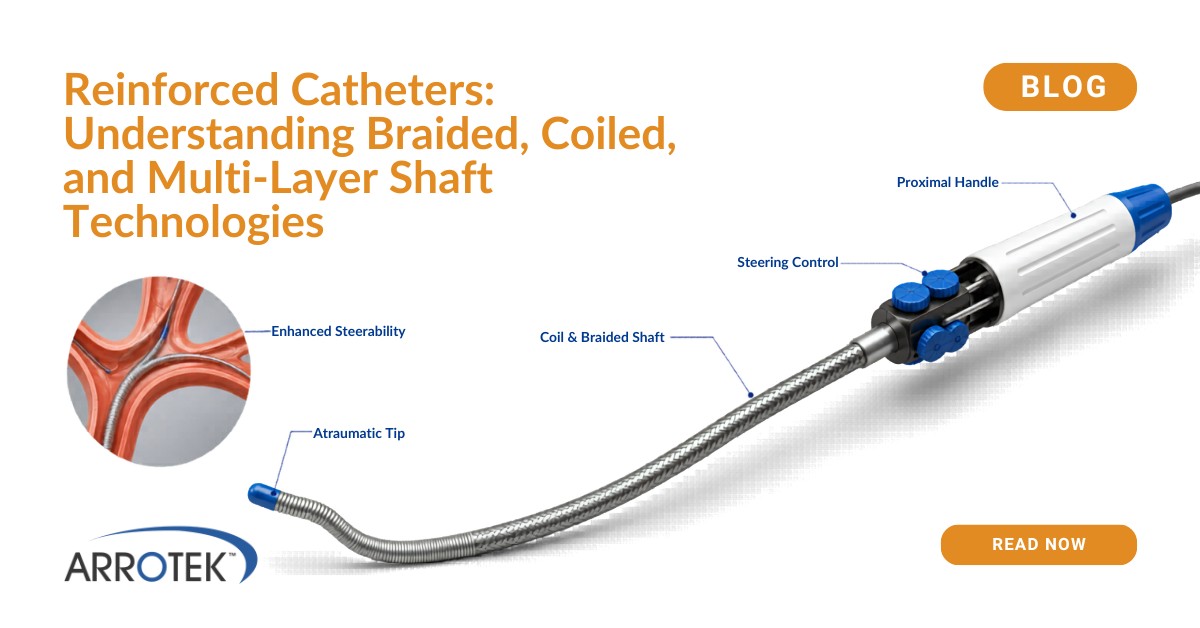
Medical device technology transfer is the process of moving from in-house manufacturing or an existing manufacturing partner to a new manufacturing partner. This includes CMOs (contract manufacturing organisations) and CDMOs (contract development and manufacturing organisations).
It is a process that is significant in both decision-making terms and execution, i.e., choosing the right CDMO partner and then ensuring the transfer takes place smoothly. In other words, this is not your run-of-the-mill switch of suppliers. Instead, it’s a complex, regulated, high-stakes project.
It’s often also an essential undertaking whether you are experiencing problems with an existing CDMO or dealing with another issue, such as capacity constraints or cost pressures. Other drivers for moving to a new CDMO, such as strategic consolidation of your manufacturing operations, are no less important.
Then there are the challenges, including those that apply during technology transfer projects in any industry. For medical device products, you also have additional regulatory compliance and validation steps to complete, in addition to quality and patient safety considerations. Get it wrong, and the consequences can be wide-ranging and severe, from supply disruption that damages commercial relationships to product quality that impacts compliance and patient safety.
On the flip side, when medical device technology transfer is done well, you can improve processes, reduce costs, enhance resilience, and deliver a better service and product to your customers. You can also open the door to new product development, R&D, partnership, and commercial opportunities.
Table of Contents
What We’ll Cover in This Guide
In this guide to medical device technology transfer when moving to a new CDMO, we will clarify what technology transfer is and the situations where it offers business and operational benefits.
We’ll explore the regulatory and quality framework that you must work within, the importance of good documentation, and how to choose the right CDMO partner.
And we’ll outline the key steps in the technology transfer process, challenges to overcome, and best practices to achieve an optimal outcome.
What Is Medical Device Technology Transfer?
Medical device technology transfer is a highly structured process for moving the knowledge, documentation, processes, equipment, and expertise required to manufacture a medical device product from one manufacturing site to another.
It can include moving machinery and always involves handing over drawings, but this is only a small part of the process. Instead, think of it as moving an entire manufacturing ecosystem – process knowledge, quality systems, supplier relationships, your standing with regulators, etc. It’s also a highly regulated process that is governed by both quality systems (such as ISO 13485) and FDA, EU MDR, and potentially other regulatory requirements. In fact, in most situations, the transfer will trigger a formal change control process in your QMS that may require regulatory notification or submission.
As a result, medical device technology transfer projects must be properly planned and carefully executed, with all steps fully documented in your quality management system (QMS).
There are three main types of medical device technology transfer, each presenting its own requirements and challenges:
- From in-house manufacturing to a CDMO – this is a documentation-heavy process that needs to be fully managed, especially if documentation is missing or held by individuals rather than systems.
- From an existing CMO/CDMO to a new CDMO – considerations with this situation include the level of cooperation offered by the existing manufacturer, IP considerations, and potential gaps in documentation.
- From an internal site to a different internal site – even though all parties are the same “team”, this situation still requires comprehensive management, full change control, and validation.
In all situations, the objective is the same, i.e., the new CDMO must be able to manufacture the product or component to the same specification and at least the same quality standard as before. Manufacturing consistency at the new CDMO is also essential, all processes must be properly validated, regulators must be updated, and your QMS must reflect the new manufacturing circumstances.
When Should You Consider Moving to a New CDMO?
Moving to a new medical device CDMO is not a decision that should be taken lightly. There will be costs involved, it can be time-consuming, and there are risks. Therefore, it’s essential to have a clear understanding of your current situation and the triggers that indicate it is time to move to a new CDMO.
Quality and Compliance Failures
Regular non-conformances, failed audits, and regulatory warning letters are examples of quality and compliance failures by CDMO partners (and sometimes even in-house manufacturing operations) that can be damaging to your brand and product reputation, the profitability of your operations, and your standing with regulators.
Capacity Constraints
Capacity constraints can arise for a range of reasons, including existing manufacturing operations (either in-house or by a CDMO) being unable to scale to keep up with your growth. This includes when moving from early-stage production for a new product to scale-up production as your commercialisation efforts ramp up.
Other examples include a CDMO prioritising other customers over your products or the CDMO being unable to consistently meet lead times.
Cost Pressures
Cost pressures are a common driver for moving to a new medical device CDMO, especially if you have small in-house manufacturing operations or are currently partnered with a smaller CDMO. These types of situations make it more difficult to spread costs or absorb price increases for things like energy, raw materials, and equipment maintenance.
Commercial Relationship Strain
Commercial relationships change over time, and some of those changes can trigger a move to a new CDMO. An example is where your existing CDMO imposes new and challenging MOQs (minimum order quantities). Other examples are where your existing CDMO is restructuring, going through a period of financial difficulty, or closing the facility responsible for manufacturing your products or components.
Strategic Consolidation
The above triggers for moving to a new CDMO are largely reactive, i.e., you are responding to a situation to make improvements. There are also proactive triggers for partnering with a new CDMO, including streamlining your supply chain, bringing manufacturing closer to your main commercial market, or moving to a CDMO that has a broader range of capabilities.
Supply Chain Resilience
Reducing supply chain risk is another proactive motivation for moving to a new CDMO, including when diversifying from a single manufacturing region or moving away from a region going through a period of instability.
Future Development Plans
As you develop new iterations of your product, you might need to move to a new CDMO that has the technical capabilities to support next-generation versions or new device features.
Key Questions to Ask
| CDMO transfer trigger | Key questions to ask |
|---|---|
| Quality & compliance failures | Are failures frequent? Is the root cause systemic or isolated? Has the issue affected a product in the market? |
| Capacity constraints | Is the capacity issue temporary or structural? Is the situation likely to change, such as through an existing CDMO investing to increase capacity? |
| Cost pressures | Have you benchmarked current pricing? Can you negotiate better terms? Are there efficiencies that can be made that will reduce costs? |
| Commercial relationship strain | Can the relationship be improved? When will your business be impacted by any changes? |
| Strategic consolidation | Does your new CDMO partner have all the technical and operational capabilities that you need? Will consolidation create new single points of failure? |
| Supply chain resilience | Can you quantify your current supply chain risks? How will changing CDMO mitigate those risks? |
| Future development plans | Can you see a gap in future capability? Are there other solutions, such as existing manufacturing operations, to improve capabilities? |
There are two final points to highlight in terms of when you should consider moving to a new CDMO. The first is to address the situation honestly, as delaying a decision will likely increase risks. The second is to highlight that once a decision to move is taken, regardless of the motivations, the approach should be structured, documented, treated as a regulated project, and closely managed – from day one.
The Regulatory and Quality Framework
As mentioned in the previous section, medical device technology transfers must be treated as regulated projects. But what is the regulatory framework you must operate within? What are the quality requirements and implications?
The main standards and regulations are in the quick reference table below. Before that, there are essential points to highlight:
- Medical device technology transfers should be managed via a change control mechanism within a formal quality system – QMS.
- The change order should document what is changing, the risk assessment, the validation activities that are required, and any necessary regulatory notifications or submissions.
- The process might have implications for the regulatory status of the product, so this should be planned for in advance.
- OEMs retain regulatory responsibility during and after the transfer, i.e., regulatory ownership or liability does not transfer to your new CDMO partner.
- A quality agreement should be put in place to clearly define the responsibilities of the OEM and CDMO across manufacturing, quality control, change control, complaint handling, and regulatory support. This agreement should be in place before the technology transfer process begins.
Quality and Regulatory Framework Quick Reference
| Regulation or standard | Jurisdiction | Relevance to medical device technology transfer |
|---|---|---|
| ISO 13485:2016 | International |
|
| FDA 21 CFR Part 820 / QMSR | United States |
|
| EU MDR | European Union |
|
| FDA Process Validation Guidance | United States |
|
| ICH Q10 | International |
|
Note: regulatory requirements change and can vary by device, market, and the specifics of the manufacturing change. The best approach is to always conduct a formal change impact assessment and seek customised regulatory advice.
Understanding the regulatory framework that you must operate within during and after the medical device technology transfer is essential for success. Not only does understanding the regulatory framework ensure compliance, but it also ensures the scope of validation is fully understood, as well as the timeline and costs that will be involved.
Key Documents Required for a Successful Transfer
The importance of documentation in successful medical device technology design processes cannot be overstated. In fact, documentation is the foundation of the process, with gaps in documentation being one of the main causes of delays, failed validations, and regulatory issues.
Given the importance of documentation, preparation should begin long before the technology transfer gets underway. The best starting point for OEMs is typically to audit the documentation package to identify gaps, outdated records, and knowledge that has never been formally captured.
Your CDMO also needs to generate documentation as part of the transfer process. These documents have different purposes, but they are no less important.
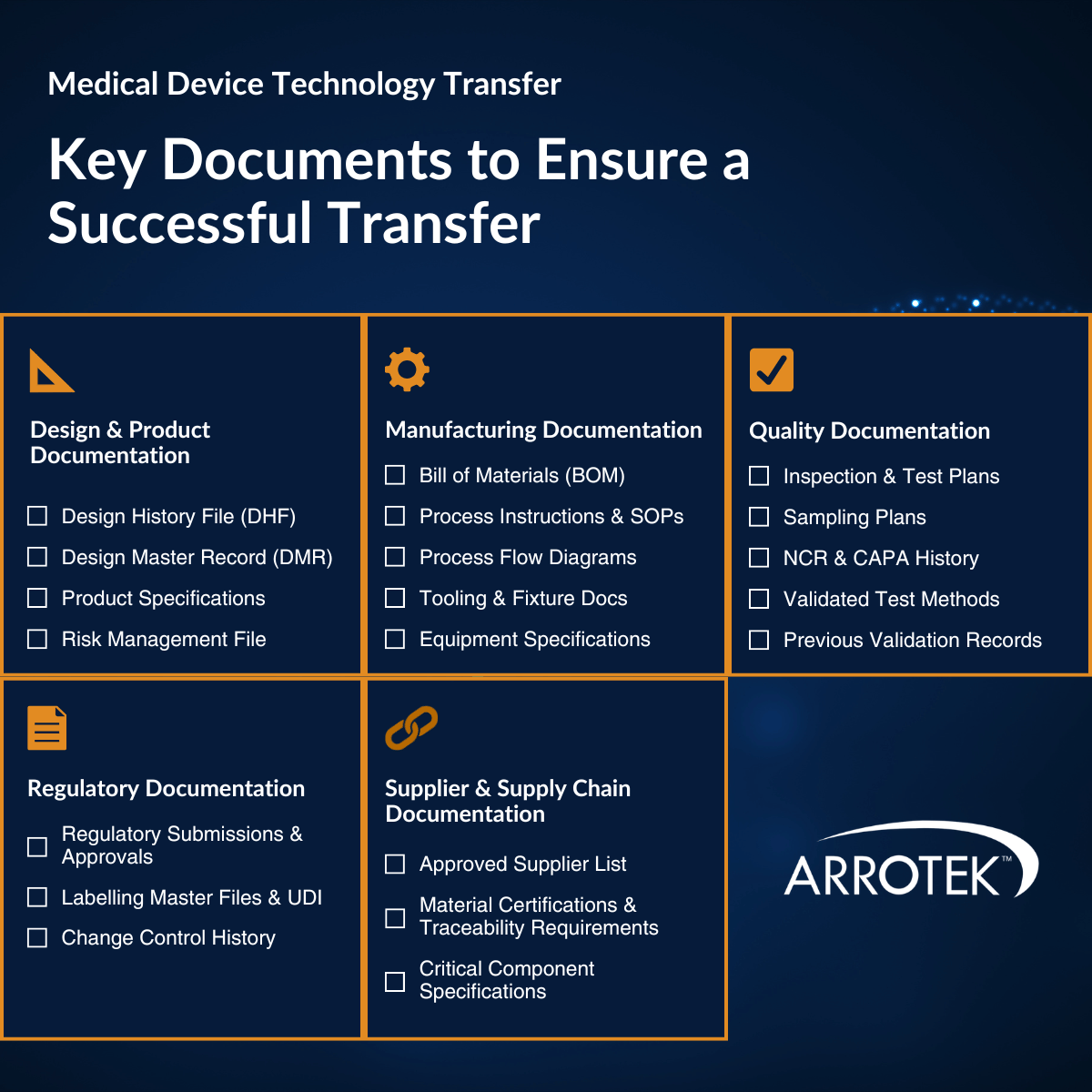
Design and Product Documentation
- Design History File (DHF) – the complete record of the design and development of the product or component.
- Device Master Record (DMR) – the main set of documents that define how the product or component is manufactured. The DMR typically includes drawings, specifications, bills of materials (BOMs), and SOPs.
- Product specifications – dimensional, material, functional, and performance specifications for the product or component.
- Risk management file – risk management plan, risk analysis documentation, and risk evaluations.
Manufacturing Documentation
- Bill of materials (BOM) – including all components, sub-assemblies, and materials. A BOM often also includes approved suppliers and part/sku numbers.
- Manufacturing process instructions and SOPs – current step-by-step instructions for each manufacturing process and operations.
- Process flow diagrams – visual maps of the manufacturing process.
- Tooling and fixture documentation – drawings, specifications, and qualification records for the tooling, jigs, and fixtures used to manufacture the product or component.
- Equipment specifications – details of the manufacturing equipment currently used to manufacture the product or component, including critical parameters, calibration parameters, and maintenance schedules.
Quality Documentation
- Inspection and test plans – defining what is inspected, when, and by what method. The acceptance criteria should also be included.
- Sampling plans – the methodology used for statistical sampling, including for raw materials, in-process sampling, and final inspections.
- Historical non-conformance and CAPA (corrective and preventative actions) records – it is often beneficial to give the new CDMO details of previous quality issues and the solutions that solved them.
- Validated test methods – especially in relation to performance testing.
- Previous validation documentation – this can provide a baseline for the new CDMO and help inform what will be required.
Regulatory Documentation
- Regulatory submissions and approvals – 510(k) clearances, CE certificates, technical files, and PMA (pre-market approval) documentation relevant to the product or component.
- Labelling master files – approved labelling (including market-specific labelling requirements) and UDI (unique device identification) information.
- Change control history – the record of previous changes to the product or component, or its manufacturing process.
Supplier and Supply Chain Documentation
- Approved supplier list (ASL) – approved suppliers and their qualification status.
- Material certifications – certificates of conformance, material traceability requirements, and any inspection criteria that are connected to specific suppliers.
- Critical component specifications – where supplier or material changes could affect the product’s performance or regulatory status.
9 Step Medical Device Technology Transfer Process
The medical device technology transfer process can be categorised into nine broad steps or phases. However, it’s important to highlight before getting into the steps the fact that many overlap, as technology transfers are not linear processes. Understanding the steps is still essential, though, as they provide a structured framework that keeps the project proactive and controlled.
It’s also beneficial to note that timelines for each of the steps can vary depending on the type of device, the complexity of the manufacturing process, and other issues such as the availability of documentation.
The nine main steps in the medical device technology transfer process include:
- Feasibility and CDMO selection
- Project planning
- Knowledge transfer
- Equipment and tooling qualification
- Process development and optimisation
- Validation
- Regulatory submission or notification
- First article and production roadmap
- Closeout and ongoing governance
Step 1: Feasibility and CDMO Selection
The starting point is to assess whether the CDMO has the capability to manufacture your medical device product or component. This process should include a capability assessment and a facility visit so you can conduct a comprehensive technical review.
Part of this process should include evaluating the CDMO’s QMS and ISO 13485 certification. Look at things like CAPA effectiveness, findings from internal audits, and how the company handles non-conformances to get a full understanding of the maturity of their quality system.
Tips to help you choose the right medical device CDMO partner include:
- Identify a CDMO partner that has direct experience manufacturing products or components that are the same as yours. Specific, direct experience is always better than general capabilities.
- Issue a detailed RFQ to ensure the CDMO fully understands what is involved and to get a meaningful response.
- Complete NDAs before you share any technical documentation.
- Ask for (and check) references from existing customers.
- Check the CDMO’s regulatory track record using your own research. This should include checking the FDA’s warning letter database and available inspection observations (Form 483s)
- Evaluate the team beyond the commercial team you are negotiating with, i.e., the production manager, quality manager, process engineer, etc.
- Assess the financial stability of the CDMO, including its ownership structure.
- Consider the often-overlooked but very important cultural fit dimension. Site visits help with this consideration.
- Don’t select a CDMO solely on price. Costs are important, but there are many other factors that also need to be taken into account.
- Agree commercial terms and a quality agreement before fully committing to the technology transfer to prevent disputes and unwelcome surprises.

Download the CDMO Evaluation Scorecard as a PDF.
Step 2: Project Planning
The cornerstone of the project planning phase is to establish a formal technology transfer plan as a controlled document within the QMS. The document should define crucial regulatory and operational areas such as the scope of the technology transfer, where responsibilities lie, the acceptance criteria, and the validation strategy.
The best approach is where both the OEM and CDMO have a dedicated project lead assigned to the technology transfer. These individuals should have minimal workload responsibilities outside the technology transfer to ensure maximum focus.
Other technology transfer project planning tips include:
- Define roles and responsibilities explicitly, as ambiguity causes delays.
- Timelines can be demanding, but it is essential that they are also realistic.
- Include milestones and decision gates in the timeline.
- Include a buffer in the timeline for the unexpected and situations that you can’t fully control, such as regulatory review cycles, equipment lead times, and documentation gaps.
- Create a risk register at the very start of the process to capture all risks – technical, supply chain, regulatory, timeline, etc.
- Establish an agreed communication cadence and method.
- Define the acceptance criteria for the technology transfer upfront so everyone understands the expectations.
Step 3: Knowledge Transfer
All stages of a medical device technology transfer carry risks, but this step is arguably the riskiest, especially in relation to invisible risks. This is because documentation can be transferred from one manufacturer to another easily. Knowledge is much more difficult to transfer.
Tips to optimise the technology transfer phase include:
- Conduct structured, deep-dive technical workshops (plural) between the existing manufacturing team and the team at the new CDMO.
- Implement a strategy to address the tacit knowledge and undocumented intellectual capital that currently exists. A key part of this strategy should be speaking to the operators on the production line directly, not just the engineers who wrote the SOPs.
- Spend time bringing the new CDMO through the non-conformance and CAPA history to ensure previous mistakes are not repeated.
- Ensure the new CDMO has a deep understanding of the clinical context of your medical device.
- Document all knowledge transfer activities.
Step 4: Equipment and Tooling Qualification
Decide on the plan for equipment early in the process. Common scenarios include:
- Equipment is transferred from the current manufacturer to the new CDMO.
- Equipment is replicated by the new CDMO.
- The CDMO uses existing equipment.
Each of the three options above has different validation implications (as well as potential logistics and cost implications). For example, a machine that is fully validated at the existing manufacturing facility is not automatically validated at the CDMO.
Additional tips to optimise the equipment and tool qualification phase include:
- Make sure all qualification processes are documented – Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ).
- Make sure calibration and maintenance systems are put in place at the new CDMO before validation begins.
- For interventional medical devices, tooling requires extra attention in terms of accurate documentation and agreed acceptance criteria.
- Do not leave areas of ambiguity in relation to the ownership of tooling.
Step 5: Process Development and Optimisation
It is inevitable that production processes at the new CDMO will differ slightly from the processes at the existing manufacturing facility. As a result, process development will almost certainly be required as part of the technology transfer. It’s also important to note that complex devices are likely to need multiple process development iterations.
Tips for process development and optimisation include:
- Run engineering or feasibility builds before committing to validation. Use the engineering or feasibility builds to identify potential issues and establish things like process parameters and preliminary settings.
- Define and formally document the relationships between critical process parameters (CPPs) and critical quality attributes (CQAs).
- Make sure the new CDMO’s quality team is involved in this part of the technology transfer process.
- Establish a positive, problem-solving culture in relation to this process. For example, view engineering builds that produce non-conforming products as a positive step in the process rather than simple failures.
Step 6: Validation
The validation phase of a medical device technology transfer project is about converting the engineering work done to date into documented and auditable evidence. Due to its complexity, it is typically the phase that takes the longest and incurs the highest costs. Working methodically and strategically through the previous steps helps to optimise validation processes and reduce both the timeline and the costs involved.
The FDA’s Process Validation Guidance is a robust and structured approach to validation in a medical device technology transfer project. It is based on a three-stage lifecycle model:
- Process design (completed in the previous step)
- Process qualification
- Continued process verification
A key document that controls the validation process is the validation master plan. It should map out all validation activities, including:
- Process validation
- Equipment qualification
- Analytical method validation
- Software validation
- Packaging validation
- Sterilisation validation
Installation Qualification (IQ)
- Confirm that the equipment is installed correctly.
- Document utilities, calibration status, and software.
- Verify maintenance and calibration schedules.
Operational Qualification (OQ)
- Confirm equipment operates within defined parameters
- Challenge the operating limits for critical process parameters.
- Carry out worst-case scenario testing.
- Document all deviations.
Performance Qualification (PQ)
- Confirm the process consistently produces conforming products under commercial manufacturing conditions.
- Define the number of PQ runs and the acceptance criteria.
- Ensure PQ batches are manufactured across different shifts and operators.
- Ensure PQ batches are representative of commercial manufacturing.
- Fully investigate and resolve non-conformances.
Analytical Method Validation
- Demonstrate that the test methods used to evaluate product conformance are fit for purpose.
- Address key validation parameters, including accuracy, precision, repeatability, reproducibility, specificity, linearity, and detection limits.
- Pay particular attention to performance test methods applicable to interventional medical devices. Examples include tensile strength, tip deflection, kink resistance, burst pressure, and flow rates.
- Ensure the acceptance criteria are agreed in advance.
Packaging Verification
- Don’t leave packaging verification to the end – plan early.
- If packaging materials, formats, or suppliers are changing, a full packaging validation programme might be required.
- Plan for key testing processes – seal integrity testing, package integrity testing, distribution testing, and accelerated ageing studies.
Sterilisation Validation
- Sterilisation validation, where required, is often time-consuming, so plan for it early in the process.
- Sterilisation method and cycle parameters will need to be validated for the new CDMO.
Software Validation
- Software used to collect quality data or control manufacturing or batch release processes must be validated – manufacturing execution systems, electronic batch record systems, etc.
- Software validation should follow a risk-based approach.
Other Technology Transfer Validation Considerations
- If materials, processes, or cleaning agents are changing at the new CDMO, a formal biocompatibility assessment might be required.
- Validation should not be considered complete until deviations are evaluated, resolved, or formally justified and approved.
- Note that the validation summary report is a crucial regulatory document.
- All validation documentation should be stored within the QMS.
Step 7: Regulatory Submission or Notification
The work completed in steps one to six will create a validated manufacturing capability at the new CDMO. Step seven turns that capability into legal market authorisation to sell the products manufactured at the new CDMO.
Step seven is an example of the non-linear characteristic of a medical device technology transfer process. While we are labelling it as step seven, planning for this phase should start much earlier in the overall process. In other words, this phase is not a post-validation activity. Preparation work should be ongoing. This especially applies if your product is sold in multiple markets, given the additional regulatory complexity this brings.
The foundation of regulatory activity during a technology transfer project is the change impact assessment. This assessment will determine what regulatory actions are required. Key elements of the assessment include evaluating whether manufacturing changes affect the safety, effectiveness, or specification conformity of the product. Every decision made during this assessment must be documented.
Step 8: First Article and Production Ramp Up
In this phase, the technology transfer process moves into the reality of commercial production. It’s important to note that this is still a transitional phase where issues can arise, so continued OEM oversight is recommended.
This part of the process includes a First Article Inspection (FAI), i.e., a formal, documented inspection of the first product manufactured at the CDMO. FAI best practices include:
- Define the acceptance criteria in advance.
- Acceptance criteria should mirror the product release criteria in the DMR.
- Both OEM and CDMO quality teams should be involved in the FAI.
- Fully document the FAI.
- Failures should trigger a formal non-conformance process.
- For complex products, consider conducting FAI across multiple batches.
Moving to production ramp-up, this should be a controlled and structured process. This includes monitoring essential KPIs – yield, non-conformance rate, first-pass inspection rate, cycle time, CAPA volume and closure rate, and on-time delivery performance.
It’s also beneficial to treat production ramp-up issues (which are likely to occur) as valuable information rather than failures.
Step 9: Closeout and Ongoing Governance
This phase marks the end of the technology transfer process, but it shouldn’t be rushed or treated as a simple administrative exercise. Completing it properly helps with the transition to a successful ongoing operational state.
Key tasks in this phase include:
- Produce a technology transfer report within the QMS.
- Formally close the change control in the QMS.
- Conduct a lessons learned review.
- Review and update the quality agreement.
- Transition to ongoing oversight and governance with periodic business reviews, KPI reporting, audit schedules, change notifications, and agreed escalation paths.
Common Challenges and How to Overcome Them
Loss of Undocumented Intellectual Capital (Often Referred to as Tribal Knowledge)
- Start capturing undocumented knowledge before starting the technology transfer.
- Engage experienced operators with interviews, work shadowing, and process walkthroughs.
Documentation Gaps and Poor Document Quality
- Complete a document audit before starting the technology transfer.
- Build documentation remediation into the technology transfer plan.
Equipment Non-Equivalence
- Complete an equipment equivalence assessment to understand the differences.
- Assess the impact of those differences on critical process parameters and critical quality attributes.
Supplier Qualification at the New CDMO
- Map out the supplier landscape, focusing especially on critical and long-lead suppliers.
- Where alternative suppliers are proposed, carefully assess the change impact.
Underestimating the Timeline
- Build the timeline based on workstream estimates rather than a target completion date to ensure the timeline is realistic.
- Add a contingency to each phase as a timeline buffer and treat the technology transfer steps as non-linear, i.e., start every activity and task as early as possible.
Best Practices for a Smooth Medical Device Technology Transfer
- Treat the technology transfer as a regulated project from day one.
- Start work to clean up, complete, and update your documentation package right away, i.e., before the transfer begins.
- Take into account the technology transfer process when considering the CDMO, i.e., does the CDMO have a proven track record?
- Plan for the reality of technology transfer processes rather than the ideal scenario.
- Implement a parallel supply strategy to mitigate risks during the production ramp-up stage at the new CDMO.
- Invest in the knowledge transfer process to ensure your highly valuable undocumented intellectual capital is not lost.
- Start packaging and sterilisation validation early.
- Regulatory planning must start early in the overall process and should be considered as a critical path activity.
- Establish clear acceptance criteria before the technology transfer work begins.
- Treat your relationship with your new CDMO as a partnership rather than a transactional relationship.
- Don’t allow short-term commercial pressures compromise transfer quality, as quality compromises can have long-term impacts.
Conclusion
A medical device technology transfer is not a manufacturing project with a bit of regulatory paperwork to complete. Instead, it is a regulated, strategic, often complex, quality-critical programme. As such, it should have the same focus as a product development project.
Approaching a technology transfer in this way brings significant benefits to medical device OEMs. The transfer itself will produce better outcomes, from improved processes to stronger supply chains. A well-managed technology transfer also supports commercial growth.
The CDMO you select is crucial to the success of not only the technology transfer but also the ongoing commercialisation of your product. This is where we come in at Arrotek, as we offer substantial CDMO capabilities at our modern, purpose-built manufacturing facility in Costa Rica. This facility specialises in high-volume, high-speed manufacturing of speciality medical needles and guidewires.
As well as manufacturing experience, our team also has extensive expertise and a proven track record in medical device technology transfer processes. By partnering with us, not only will you gain a manufacturing partner that can help you achieve your commercial goals, but we’ll work with your team to ensure the technology transfer process runs as smoothly as possible. Get in touch to find out more and to arrange a consultation.